La comparación de secuencias y estructuras desveló que proteínas semejantes a nivel de su secuencia
tienen también semejantes estructuras terciarias. Sabemos además que proteínas relacionadas evolutivamente
pueden mantener su semejanza estructural aún perdiendo su semejanza a nivel de secuencia.
Para expresar la semejanza a nivel se secuencia se usa normalmente el % de identidad que mide
la proporción de posiciones de un alineamiento que son idénticas entre las secuencias alineadas. Hay
otras formas más complicadas de expresar este concepto, pero sólo mencionaremos el e-value, una
medida que hace referencia a bases de datos de secuencias. Si esta base contiene 1000 secuencias y
las alineamos contra una secuencia X, cada una de ellas tendrá asociada un e-value para su
alineamiento con X, que será el número esperado de secuencias en la base que obtendrian la misma
puntuación por puro azar. De esta manera alineamientos con e-values bajos serán significativos.
Para expresar la semejanza estructural se usan también muchas formas de medida, pero casi todas ellas
se basan en el RMSD, la raíz de la desviación cuadrática media de las posiciones de pares de átomos de
dos proteínas que queremos comparar. Es una función sencilla de calcular a partir del alineamiento o
superposición estructural de 2 proteínas ![]() and
and ![]() , donde
, donde ![]() es el número de residuos alineados,
normalmente representados por sus
es el número de residuos alineados,
normalmente representados por sus ![]() o
o ![]() , y
, y ![]() es la distancia entre dos residuos:
es la distancia entre dos residuos:
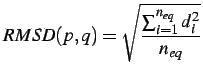 |
(1.1) |
Chothia & Lesk (1986) cuantificaron por primera vez este principio de secuencias similares
tienen estructuras similares en un número relativamente pequeño de pares de proteínas
relacionadas evolutivamente. Para cada par definieron el corazón (core) del plegamiento como la fracción
de residuos que podían superponerse a menos de 3Å of RMSD, usando las coordenadas de sus ![]() .
Con los datos obtenidos pudieron ajustar una función exponencial que relaciona la semejanza secuencial
con la estructural en el corazón del plegamiento:
.
Con los datos obtenidos pudieron ajustar una función exponencial que relaciona la semejanza secuencial
con la estructural en el corazón del plegamiento:
La figura 1.8 muestra unos datos equivalentes a los de Chothia y Lesk obtenidos con un número mucho mayor de pares de roteínas. Incluye datos obtenidos de un experimento de evaluación continua de servidores de MCP, EVA (Eyrich et al., 2001), que muestran como la curva de Chothia y Lesk limita las aplicaciones del MCP. En general se puede decir que los servidores de MCP construyen modelos con errores a nivel del esqueleto peptídico de 1Å si el % de identidad está en torno al 95%. Si la identidad baja al 30% el RMSD esperado es ya de 4Å. Además, en la figura se ve que al bajar la identidad de secuencia es más difícil predecir el error cometido.
![\begin{figure}
\begin{center}
\includegraphics[width=0.7\textwidth]{EVA_CM_bench}
\end{center}
\end{figure}](img23.png) |
Aparte de estas limitaciones intrínsecas de las técnicas de MCP, los errores en la elección de moldes
y sus alineamientos afectan negativamente a los modelos obtenidos. Por otro lado, de momento no hay
herramientas fiables para calcular el arreglo de más de un dominio, es decir, para predecir estructuras
cuaternarias, ni para predecir de manera ordinaria la estructura ni la dinámica de dominios flexibles.
Por último, la calidad de los los modelos comparativos de proteínas sólo puede evaluarse a posteriori,
porque a diferencia de las técnicas experimentales normalmente no intervienen en su construcción datos
obtenido en el laboratorio que sirvan de control.
Talvez ahora podremos apreciar mejor la figura de las aplicaciones del MCP 1.1.
